
Molecular basis of neurocognitive dysfunction and psychosis in Alpha-Mannosidosis
 0
0
Abstract
A significant portion of patients who are afflicted with lysosomal storage diseases (LSDs) encounter neurological manifestations, including cognitive issues and developmental delay, seizures, psychiatric issues, and an overall neurodegenerative decline. In order to enhance the development of effective therapies for these symptoms, it is imperative that we allude to the neuropathophysiology that underlies these manifestations. These distinct neurological and developmental features are particularly evident in patients with Alpha-Mannosidosis (AM), a type of LSD. However, there is limited published information regarding the mechanisms and pathophysiology of these presentations in patients with this condition. Although the precise impact of lysosomal storage on the biogenesis and functioning of neuronal cells has not been clearly defined, recent studies have placed emphasis on the significance of synaptic defects influencing this dysfunction. These defects encompass changes in synaptic spines, proteins, and vesicles, as well as postsynaptic densities that potentially precipitate functional disruptions in synaptic transmission and neurodegeneration. Ultimately, this cascade is thought to result in extensive neuronal loss and, consequently, the onset of cognitive manifestations. Uncovering the effects on synaptic components in LSDs with neurological symptoms like AM will enable a better understanding of disease progression. It will also allow us to identify critical targets for therapeutic intervention and the determination of optimal time frames for targeted treatments, as well as the effects of these treatments on mitochondrial function. The available therapeutic modalities in AM are not a definitive cure for affected patients, but rather an attempt to reduce the symptomatic severity in their presentation, while aiming to regress/slow down disease progression. This review will aim to discuss and rationalize the current treatment approaches in place for AM patients in relation to their effects on the improvement of neurocognitive symptoms in affected AM individuals.
Keywords
INTRODUCTION
Lysosomal storage disorders (LSDs) are a subgroup of metabolic disorders that can be characterized by a range of deficiencies of different components within lysosomal function. Specific LSDs themselves are individually rare, but collectively are one of the most prevalent groups of metabolic diseases and have been estimated to affect approximately 1 in 5,000 live births[1]. These inherited diseases stem from lysosomal gene defects, resulting in deficient or dysfunctional lysosomal enzymes, activator proteins, or transmembrane proteins[2]. This leads to the unwanted accumulation of biomolecules within the lysosome. This excess storage of macromolecules is thought to start during the stages of early embryonic development, but the clinical presentation of LSDs is highly variable, ranging from mild to severe phenotypes that may have either early or late onset[3]. It is the accumulation of undigested biomolecules within the lysosome that causes disruption to normal cellular function, resulting in the symptomatic presentation of LSDs, including neurocognitive impairment, worsening developmental delay, and abnormal neurological symptoms.
The involvement of the Central Nervous System (CNS) within this group of disorders is highly prevalent and these neurological symptoms most commonly have a slow and subtle progression. This is observed in the autosomal recessive LSDs such as Alpha-Mannosidosis (AM). The condition is a highly rare, progressive LSD that is estimated to affect 1:250,000 to 1:1,000,000 live births[4]. AM is caused by a deficiency in lysosomal alpha-mannosidase enzyme activity, consequent of mutations present in the MAN2B1 gene on chromosome 19 (19p13.13)[5]. This deficiency of alpha-mannosidase activity affects the degradation of glycoproteins, resulting in the accumulation of intracellular mannose-rich oligosaccharides in various tissues and organs, thus leading to defective cellular functions and apoptotic mechanisms[5]. AM is an abundantly heterogeneous condition that is defined by a wide variety of presentations regarding disease severity and progression, clinical manifestations, and genetic mutations.
There are currently 183 different disease-causing pathogenic variants of the MAN2B1 gene that have been identified[6]. However, AM is a highly heterogeneous condition with no clearly defined genotype-phenotype correlation[5], and despite having distinguished clinical subtypes[7], it is difficult to predict disease progression in patients with AM. Three clinical subtypes have been reported: a mild form (type 1) recognized after 10 years of age with the absence of skeletal abnormalities, myopathy, and slow progression [Figure 1], a moderate form (type 2) recognized before 10 years of age with the presence of skeletal abnormalities, myopathy, and slow progression, and a severe form (type 3) manifested as prenatal loss or early death from progressive CNS involvement or infection[5]. However, the disease manifestations of AM display a continuum of clinical severity; therefore, this classification into three phenotypic subtypes is now rarely used[8].

Figure 1. Clinical manifestations in Alpha-Mannosidosis. The figure shows the location of the clinical manifestations of AM within the human body. Cognitive function impairment and psychosis are features affecting all these patients.
It has been hypothesized that secondary mitochondrial dysfunction may contribute to the progression of neurological symptoms of AM, such as myopathy, white matter changes [Figure 2], psychosis, or seizures. Secondary mitochondrial dysfunction may arise in these patients, consequent on their defective lysosomal function, which has been observed in several other LSDs[9,10]. Secondary mitochondrial dysfunction, in addition to endoplasmic reticulum (ER) stress, can lead to reactive oxygen species (ROS) accumulation. ROS molecules contain a minimum of one oxygen atom and one or more unpaired electrons that are produced in small quantities under normal physiological conditions[11]. However, when produced in excess, they cause oxidative stress to occur within the cell, in turn leading to cellular damage and has been shown to have particular adverse effects on the nervous system[11].

Figure 2. MRI brain scans of an adult patient with Alpha-Mannosidosis. The MRI scans depicted in the figures show subtle diffuse supratentorial deep white matter changes, as well as cerebellar atrophy within the brain of a 32-year-old untreated patient with AM, who has mild learning difficulties and new-onset psychosis.
The available therapies for AM include enzyme replacement therapies (ERT) and hematopoietic stem cell transplantation (HSCT), although the latter may prove to be more effective when administered at a younger age before the disease has significantly progressed[12]. Therefore, the potential new therapies for this condition may therefore be developed in the context of the patient’s clinical symptoms consequent to secondary mitochondrial dysfunction. This review aims to discuss the theorized mechanisms of secondary mitochondrial dysfunction in AM, and the potential neurocognitive repercussions of this defect, as well as the potential therapies for AM in the context of its clinical manifestations.
CLINICAL MANIFESTATIONS OF ALPHA-MANNOSIDOSIS
The presentation of AM is highly diverse and features a wide range of clinical manifestations such as cognitive impairment, skeletal abnormalities, immunodeficiency, hearing impairment, and coarse facial features[13] [Figure 1]. The majority of individuals affected with AM appear to be clinically normal at birth, and start to present at a young age, with their symptoms gradually worsening over time[5]. The disease phenotypic presentation also ranges from mild to severe and there are no distinct clinical phenotypes of the disease due to its wide heterogeneity.
Primary CNS disease is expected in these patients, with neurological symptomatology including poor coordination, ataxic gait, metabolic myopathy, spastic paraplegia spasticity, rigidity, dyskinesia, slight strabismus, hydrocephalus, and sensorineural deafness[14]. It is also common for patients to experience psychiatric symptoms that predominantly present from puberty into adolescence. These CNS-related symptoms observed in AM will be discussed in more detail.
Ataxia
Ataxia is the most characteristic clinical manifestation of AM. It describes the impaired coordination of the patient, stemming partially from cerebellar atrophy and demyelination of the brain[15], thus affecting areas that are responsible for muscular coordination and fine motor function. Ataxia has been described as predominantly presenting in the second decade of life[16], but can also present in smaller children, generally when they learn to walk, in which affected children appear to learn to walk somewhat later[17]. Initial signs include general clumsiness and ataxic gait, with these symptoms often appearing to worsen progressively in follow-up observations as the patient ages. The cause of the ataxia is multifactorial, with myopathy, joint involvement, and cerebellar changes being the main contributors[18]. Patients require support while walking and gradually become dependent on walking aids. Moreover, their coordination is compromised, significantly impacting their quality of life.
Myopathy
Myopathic symptoms describe the development of muscular weakness in affected patients, as well as stiffness, cramps, and spasms. In patients with AM, the progression of myopathies contributes toward their progressing ataxia. It has been shown that the muscular strength of AM patients slowly deteriorates during the first decade of life and thereafter[17]. Therefore, their motor function was gradually impaired as they aged, and their ataxic manifestations worsened. Muscular hypotonia is also common, as well as spastic paraplegia[19], which are consequences of the slow progression of muscle fiber degeneration. The data on muscle pathology in AM are limited, but we have learned that vacuoles that can be found in the muscle cells are identical to those observed in lymphocytes and other cells. They contribute to muscle fiber pathology[19]. Reticulofibrillar material or lucent space with sparse granules has been described in muscle tissue from pediatric AM cases[20]; these changes were not convincing for the authors to be able to explain the muscle weakness. The authors did not comment on mitochondrial changes.
To that end, myopathic changes resulting from secondary mitochondrial dysfunction have been observed in muscle biopsies from adults with Fabry and Pompe diseases[7]. We can therefore presume that this mechanism is present in untreated adult patients with AM as well.
Neurocognitive dysfunction
Neurocognitive impairment is observed in almost all AM patients, with varying severity amongst the different symptoms, although their early psychomotor development may appear normal. It has been suggested that intellectual disability is slowly progressing in AM patients[21], whereas others have reported that disease progression halts during puberty in these individuals[22]. Characteristic symptoms that may begin to appear first include delayed speech development and motor or mental functions[5]. Affected individuals may not initiate speech until their second decade of life and may also have a restricted vocabulary and may be difficult to understand due to their poor pronunciation. These defects in speech may potentially be the result of the patients’ congenital or later-onset hearing loss[5]. This combination of poorly developed speech and sensorineural hearing loss means that patients generally achieve better scores in non-verbal tests. However, it is very difficult to measure total mental performance in these patients, particularly children, where they undergo neurodevelopmental assessment in general intelligence, language, and visual-spatial skills, as well as their overall adaptive abilities. Conversely, patients with adult-onset disease are predominantly mild to moderately intellectually disabled, with an IQ of 60-80[23]. It has been shown that this decline in IQ occurs more drastically in the first decade of life in comparison to the second decade[24] and depends on the severity of the disease. Although other studies report positive cognitive development until the ages of 10 to 12 years, there is little development thereafter[17]. Despite this, patients with AM still present with a neurocognitive function that is increasingly underdeveloped compared to other non-affected individuals within the same age group. Behavioral problems, self-harm, emotional instability, and frustration due to inability to communicate are common features observed in adults with AM (personal observations).
Psychosis
Signs of psychiatric disorder tend to appear in the second decade of life in patients with AM[13], usually from late puberty to early adolescence. However, these psychiatric symptoms can often be missed or overlooked, particularly in patients with AM who are intellectually disabled. It has been reported that psychiatric symptoms and episodes of psychosis are present in more than 25% of adults affected with AM[25]. Despite efforts to evaluate the psychiatric syndromes in these patients, this study did not reveal that there was an underlying cause. Another study has described that periods of psychosis become more pronounced with age, rising from 33% in patients aged 11 to 20 years, up to 64% amongst patients aged 21 to 30 years[17]. These episodes may be recurrent and short in duration, usually lasting 3 to 12 weeks, but medication may sometimes be necessary to improve the patients’ symptoms. Psychosis in affected individuals may be preceded by a psychological stressor, inducing states of anxiety, confusion, delusions, hallucinations, and sometimes depression which precipitates a severe loss of function, such as decreased appetite and weight loss, or incontinence of both urine and feces[5]. These episodes may then be followed by a longer period of hypersomnia, as well as diminishment in their general ability, such as being unable to read or having difficulty speaking. When assessing individuals with inborn errors of metabolism (IEMs) such as AM, clinicians should be vigilant in recognizing atypical psychiatric symptoms due to targeted treatments generally being more effective in the early stages of psychosis, before irreversible neurological damage has occurred[25]. Importantly, the neurocognitive dysfunction and resulting psychosis occur in adulthood irrespective of any ERT, which does not cross the blood-brain barrier, or HSCT, which patients often undergo in childhood[26].
Psychiatric symptoms distinct from intellectual disability may impact 25% or more of individuals with untreated AM. Typically, these symptoms manifest from late puberty to early adolescence, with psychosis seeming to be a more common feature of adult individuals with AM[27]. Recurrent episodes of limited duration may occur in these patients, and medication may be required in order to alleviate these symptoms. In nine individuals with AM and psychiatric symptoms, the rapid onset of confusion, delusions, hallucinations, anxiety, and depression occurred following a physical or psychological stressor[28]. This led to a significant loss of function, typically lasting 3 to 12 weeks, followed by a subsequent period of somnolence, asthenia, and prolonged sleep[28]. Among the nine individuals, four underwent evaluation for the psychiatric syndrome, but no underlying cause was identified[28].
Neuroimaging
Brain MRI in untreated individuals with AM reveals evidence of cerebellar atrophy, a partially empty sella turcica, and white matter signal modifications. Progressive cortico-subcortical atrophy, especially in the cerebellar vermis, has been described[29]. High signal abnormalities involving the parieto-occipital white matter are identified on axial T2-weighted scans in some individuals and may be associated with demyelination and gliosis as described by Dietemann et al. in 1990[15].
Upon the analysis of MRI brain images of 13 patients with AM, Majovska et al. concluded that white matter changes and cerebellar atrophy are proposed to be the characteristic brain MRI features in this condition
Malaquias et al. have observed hyperintensities on symmetrical T2-weighted images or superior aspects of both thalami and dentate nuclei or cerebellum. However, this research group did not observe cerebellar atrophy and periventricular white matter hyperintensity on T2-FLAIR sequence was only mild[31].
In addition, Borgwardt et al. have demonstrated in a large cohort of 97 patients that the combination of MRS/MRI changes, elevated concentrations of cerebrospinal fluid (CSF) biomarkers, and CSF-oligosaccharides suggests gliosis and reduced myelination, as part of the CNS pathology in AM[32].
Leukodystrophy is a feature of other LSDs as well as mitochondrial diseases, and given that secondary mitochondrial dysfunction and autophagy have been postulated as a cause of lysosomal dysfunction, it is postulated that they are also responsible for neurodegenerative dysfunction and psychosis in AM [Table 1].
A summary description of neurodegenerative mechanisms in LSDs
| Lysosomal storage disorder | Clinical manifestation of neurodegenerative dysfunction in humans | Pathomechanism of CNS involvement | Reference |
| Mucopolysaccharidosis | Neuroinflammation | [33-35] | |
| I | Mental retardation (variable); skeletal, heart, respiratory, corneal abnormalities | Oxidative stress (animal models and human blood) | [36,37] |
| II | As above; cognitive impairment | Oxidative stress (human blood) Accumulation of heparan sulfate and dermatan sulfate in cerebrospinal fluid Increased cathepsin B activity in the brain tissue leads to leakage of this enzyme from the lysosome into the cytoplasm in a MPSII neuronal cell line, which in turn activates the inflammasome pathway (mouse model); Defects in the DCC-regulated signaling pathway, lysosomal acidification, and Rab7 protein levels in neurons, affecting early stages of development, as well as abnormalities in glial fibrillary acid protein (GFAP) levels, suggesting early activation of glial cells (zebrafish model); Undigested GAGs in the extracellular matrix lead to the impairment of the integrin involved in signaling axon guidance and vesicular pathways that integrate neural circuit development in the early stages of neuronal development | [38] [39] [40] [41] [42] [43] |
| III A | Developmental delay, behavioral disturbances, hyperactivity; speech delay, intellectual disability | Reduced excitatory synaptic strength on the somatosensory cortex (mouse model); inhibition of soluble NSF attachment receptor (SNARE) complex assembly and synaptic vesicle recycling, possibly caused by perikaryal accumulation of insoluble α-synuclein and increased proteasomal degradation of cysteine string protein α, resulting in low availability of these proteins at the synaptic terminal; HS accumulation in the brain causes changes in oligodendrocyte cell state (zebrafish model) | [44] [45] [46] [47] |
| III B | Developmental delay, behavioral disturbances; speech delay, intellectual disability | Oxidative stress (animal models); Golgi involvement; HS accumulation in the brain causes changes in oligodendrocyte cell state (zebrafish model); Time-dependent accumulation of HS and HS-NRE and progressive increase in LAMP1 staining in the forebrain and cerebellum leads to lysosomal enlargement, atrophy of white matter, loss of Purkinje neurons, and progressive increase in microgliosis (cortex and cerebellum) and astrogliosis (cerebellum). Raised Iba1 is an indicator of microglial activation (Canine model); Early vacuolation of glial cells and vacuolation of neurons, along with increased glial expression of GFAP and Iba1, suggest neuroinflammation occurs early on in disease within the spinal cord and dorsal root ganglion (DRG) (Canine model) | [48,49] [46] [47] [50] [51] |
| III C | Developmental delay, behavioral disturbances, hyperactivity; speech delay, intellectual disability | The primary accumulation of HS in microglial cells and neurons results in impaired autophagy, leading to secondary neuronal storage of GM2/GM3 gangliosides and misfolded proteins, neuroinflammation, and abnormalities in mitochondrial energy metabolism. This cascade of events ultimately leads to neuronal death. Within the terminals of MPSIIIC hippocampal neurons, synaptic vesicles are reduced; these neurons exhibit changes in distribution of excitatory synaptic markers and in transmission. There is a progressive deficiency in mitochondrial function, with a selective reduction in OXPHOS complexes and decreased coenzyme Q10. HS accumulation in the brain causes changes in oligodendrocyte cell state (zebrafish model) Significant decrease in the frequency and amplitude of excitatory and inhibitory miniature synaptic events (mEPSCs and mIPSCs) (mouse model) | [52] [53] [54] [55] [46] [47] [54] |
| Multiple sulphatase deficiency | Neurological deterioration, ichthyosis, skeletal anomalies, and organomegaly | Autophagy and mitophagy accumulation fragmentation decreased ATP content | [55] [56] |
| Niemann-pick | |||
| A/B | Severe deterioration of the central nervous system (CNS), accompanied by the storage of sphingomyelin in both visceral and cerebral regions | TRPML1-mediated Ca2+-release is compromised | [57] [58] |
| C | Sub-acute nervous system involvement, characterized by a relatively moderate progression and less prominent visceral storage pathology; cerebellar ataxia, dysarthria, dysphagia, progressive dementia, and occasionally seizures; progressive neurological regression, seizures, spasticity | Synaptic pathology (mouse model); impairment of SNARE function; mitochondrial cholesterol accumulation; the simultaneous primary storage of cholesterol, coupled with the secondary storage of sphingomyelin, is a prime driver for NPC1 pathology, interfering with TRPML1 and TRPML1-dependent maintenance of lysosomal homeostasis. Sphingolipids show mislocalization from the Golgi apparatus to lysosomes, as demonstrated by impaired trafficking of lactosylceramide in NPC1 cells. Abnormal lipid accumulation in NPC1 patient lysosomes results in secondary lysosomal storage by blocking TRPML1- and Ca2+-dependent lysosomal trafficking; this storage could be reverted by the TRPML agonist ML-SA1. Sphingosine storage induces calcium depletion in lysosomes, possibly through an inhibitory effect on Na+/Ca2+ exchangers. Neuroinflammation; Peroxisomal dysfunction; Decreased oxidative respiration/reduced ATP levels; Increased vulnerability to oxidative stress; Decrease in mitochondrial GSH resulting in Cytochrome c release | [59] [60] [61] [62] [63] [61] [63] [64] [65,66] [67] [68] |
| Gaucher disease I-III | Progressive neurological regression, seizures, spasticity (mainly III), cognitive impairment, psychiatric disturbances (GD I and III) | Neuroinflammation, dysregulated calcium homeostasis, decreased mitochondrial membrane potential, selective reduction of OXPHOS complexes, accumulation of APP and α-synuclein, Reduced O2 consumption/reduced ATP levels Significant systemic oxidative stress demonstrated by altered GSH status and lowered catalase enzymatic activity, as well as elevated lipid peroxidation (GD I) (human) Increased Tmem119 mRNA expression leads to abnormal microglia growth and proliferation, as well as increased Iba1 expression exhibiting a higher degree of microglial cell activation; correlated by increases in pro-inflammatory cytokines Tnfa, Il1b, and Il6, as well as chemokines Cxcl10, Ccl2, Ccl5, and Cxcl9. Markedly raised antigen-presenting cell receptor Cd86, a cell surface protein identifying M1 microglia (adult genetic nGD model) The active, phosphorylated form of c-Abl is increased, and interacts with RIPK3, which is in turn phosphorylated at a tyrosine site, and RIPK3 phosphorylation is reduced when c-Abl is inhibited. This shows that c-Abl signaling is the upstream pathway that activates RIPK3 in GD (adult human and mice GD models) Accumulating Β-glucosylceramide in GD activated microglia through macrophage-inducible C-type lectin induces phagocytosis of living neurons, exacerbating symptoms of GD. This is augmented by tumor necrosis factor (TNF) secreted from activated microglia that sensitizes neurons for phagocytosis (human model) Deleterious effects of Wnt/β-catenin downregulation in neuronopathic GD may be ameliorated by the prevention of Dkk1 binding to the Wnt co-receptor LRP6 (mouse model) In the presence of GBA1 mutations, increased chaperone and LONP1 activity may promote the degradation of damaged/misfolded intramitochondrial GCase. Complex I activity is ameliorated with LONP1 inhibition in GBA1 mutant HEK cells, with the opposite being observed in WT-Gcase cells. This is also observed in iPSC neurons, showing that in the presence of mutant GCase, LONP1 proteolytic activity is more pronounced than its chaperone function. This increased interaction between mutant GCase and LONP1 may also interfere with the folding properties of LONP1, leading to mitochondrial protein aggregation, which may result in decreased CI activity and mitochondrial α-synuclein accumulation in iPSC-derived neurons | [64] [65,66] [58] [69] [70] [71] [72] [73] [74] [75] [76] [77] [78] |
| Krabbe disease | Developmental delay, peripheral neuropathy, hearing/visual impairment, seizures | Synaptic pathology (mouse model) Peroxisomal dysfunction - downregulates the peroxisome proliferator-activated receptor-alpha (PPAR-alpha). Decreased mitochondrial membrane potential oxidative stress/GSH Dysregulation of Ca2+ signaling Cytochrome c release GALC deletion causes growth and motor coordination defects, and inflammatory gliosis due to the significant accumulation of psychosine in the nervous system. This was shown to result in profound neuro-axonal degeneration with a mild effect on myelin structure (mouse model) α-synuclein aggregation in the brain tissue to form fibrils (human) | [79] [80,81] [82] [83] [84] [85] |
| GM1 gangliosidosis | Progressive neurological regression, seizures, spasticity | Synaptic pathology (feline model) Neuroinflammation; enhanced autophagy and mitochondrial dysfunction. Dysregulated calcium signaling. Decreased mitochondrial membrane potential. Increased vulnerability to oxidative stress-induced Cytochrome c release Microglial activation with increased levels of LC3 autophagy regulator; increased microglial activation and proliferation in the cerebral cortex (mouse model) | [86,87] [88] [89] [90] [91] |
| GM2 gangliosidosis (Tay Sachs) | Progressive neurological regression, seizures, spasticity, deceleration in movement, ataxia, tremors; psychosis | Dysregulated ER calcium homeostasis Neuroinflammation; the production of inflammatory mediators, cytokines and chemokines, Ccl2, Ccl3, Ccl4, Cxcl10, Cxcl13. Ccl2 displays chemotactic activity for monocytes, lymphocytes, and neutrophils. Ccl3 influences monocyte, lymphocyte, and neutrophil migration together with Ccl2, and activation of T cells and macrophages with Ccl4. Cxcl10 produced by astrocytes recruits activated T lymphocytes by increasing their migration to the site of tissue damage in the cortex and cerebellum. This leads to cytoplasmic vacuolation of nerve cells, deterioration of Purkinje cells, and neuronal death, preceded by activated microglia expansion, macrophage and astrocyte activation (mouse model) | [58] [92] [93] |
| Mucolipidosis IV | Mental impairment, speech impairment, spasticity, neuroaxonal dystrophy, blindness; intellectual disability | Lack of the endolysosomal ion channel mucolipin1/ TRPML1/MCOLN1, with evidence of lysosomal accumulation of gangliosides and heavy metals such as zinc and iron; Ca2+ abnormalities; secondary mitochondrial dysfunction Changes in cytokine release (IFN-α1, IP-10) in response to TRPML1 loss of function and upregulation of interferon-gamma signaling results in defective brain myelination, oligodendrocyte dysfunction and pro-inflammatory activation of microglia and astrocytes Significant delays in expression of mature oligodendrocyte markers Mag, Mbp, and Mobp in the cortex early in life result in hypomyelination and diminished oligodendrocyte maturation between the cortex/forebrain and cerebellum (mouse model) Mitochondria-lysosome contacts facilitate the direct transfer of Ca2+ from lysosomes into the mitochondria, mediated by TRPML1 which is disrupted in IV | [94] [7,95] [96] [97] [98] |
| Fabry disease | Progressive motor and nonmotor neurodegeneration; it remains unclear whether these are associated with Parkinsonian neurodegeneration | Disrupt the autophagy-lysosomal pathway, leading to autophagosomal accumulation of phosphorylated a-synuclein in the mouse brain TRPML1-mediated Ca2+-release is compromised | [7,99] [57] |
| Neuronal ceroid lipofuscinoses | Dementia, motor disturbances, epilepsy, loss of vision, and early death. Cognitive decline and seizures. | Neuroinflammation. Mitochondrial dysfunction. Reduced ATP levels. Deficient mitochondrial Ca2+ buffering. Mitochondrial vacuolation Cysteine string protein alpha (CSPα) mislocalizes as aggregates to the neuronal soma instead of being targeted to the presynapse. This results in the decreased interaction between CSPα and SNAP-25, causing increased SNAP-25 degradation and impaired synaptic SNARE-complex assembly Failed autophagy results in accumulation of impaired neuronal mitochondria; mROS accumulation is observed in Cln7 neurons that mediate glycolytic enzyme PFKFB3 activation (mouse model) CLN8 deficiency decreases the complexity and size of the somatodendritic compartment, leading to neurodegeneration (rat model) CLN5 is a substrate of CRL3-KCTD7 E3s. In NCL, KCTD7 mutations result in the disruption of the interaction between KCTD7-CUL3 or KCTD7-CLN5, leading to excessive CLN5 accumulation in the endoplasmic reticulum. CLN5 accumulation disrupts the CLN6-CLN8 interaction and lysosomal enzymes, which causes the impaired trafficking of ER-to-Golgi lysosomal enzymes. The C128Y mutation causes abnormal palmitoylation of CSPα and aggregates formation, also triggering lipofuscin deposits in adult-onset NCL (human) | [100] [101] [102] [103] [104] [105] [106] [107] [108] |
| Pompe disease | Limb–girdle muscle weakness. Intellectual disability, impaired visuospatial functioning. | Mitochondrial calcium excess, increased ROS generation, decreased mitochondrial membrane potential, and decreased oxygen consumption and ATP production | [7] [109] [110] |
POTENTIAL MECHANISMS OF NEURODEGENERATION IN ALPHA-MANNOSIDOSIS
Current therapies in AM and psychosis
The current therapeutic options for the treatment of AM include ERT with velmanase alfa and allogeneic HSCT, as well as best supportive care that addresses symptoms as they arise [Table 2]. However, there is still a lack of studies on the monitoring of treatment response and associated complications outside of clinical trials and published case reports[115]. HSCT is an effective therapy commonly used in the treatment of several metabolic diseases with associated neurological dysfunction. Post-HSCT treatment, the normal donor stem cells are able to differentiate into different cell lineages, which can colonize a range of organs and tissues. These differentiated cells have the capacity to secrete the normally functioning enzyme that is otherwise deficient or dysfunctional in the patient, which is then widely distributed due to cell-to-cell contact amongst the transplanted cells[116]. This was first tested in the treatment of AM in cat models, in which it was shown that HSCT has the ability to significantly increase the levels of alpha-mannosidase within the neuronal cells of the CNS[25]. There have since been further trials into the effectiveness of HSCT in patients with AM, with one study showing that patients who had undergone HSCT had a significantly higher recorded IQ post-treatment, as well as improved overall cognitive function, compared to patients who had not received HSCT therapy[117]. Patients who received HSCT also showed no further decline in their IQ compared to untreated individuals. HSCT has also been shown to improve intellectual function in four patients with AM, as well as improving their adaptive skills and verbal memory function in all patients[118]. All four patients also demonstrated improved speech and hearing, with no new skeletal abnormalities reported. A normalization in leukocyte alpha-mannosidase enzymatic activity was also observed in all four patients post-HSCT. HSCT has more recently been shown to improve the clinical symptoms in patients with AM, mainly with intermediate or severe disease and symptoms of neurodevelopmental delay, by Mynarek[119], which is also the largest retrospective analysis of HSCT treatment in patients with AM. It was reported that the mortality and morbidity of patients were improved, in addition to developmental improvement observed in all patients. Some patients also displayed improved hearing ability and preservation of neurocognitive function.
A summary description of neurodegenerative mechanisms in Alpha-Mannosidosis
| Lysosomal storage disorder | Clinical manifestation of neurodegenerative dysfunction in humans | Pathomechanism of CNS involvement | Reference |
| Alpha-Mannosidosis | Cerebellar dysfunction, developmental delay, absent tendon reflexes, spasticity, developmental delay, impaired speech, hearing impairment, psychosis, cognitive impairment Impaired visual function, reasoning, visuo-spatial skills, memory, and attention | Sural nerve biopsies showed neuropathy with myelin degeneration and metachromatic deposits CNS pathology of reduced myelination with elevated mannose complexes and gliosis, increased CSF-oligosaccharides. Significant elevations in Cho/Cr and ml/Cr reflect reduced myelination and gliosis. Raised NFLp concentrations due to axonal injury, degeneration, and myelin loss Iron deposits at the basal ganglia Dilated endoplasmic reticulum, increased levels of aberrant mitochondria with reduced mitochondrial mass; oxidative stress with increased ROS | [27] [111] [112] [18] [113] [114] |
These improvements in neurological function post-HSCT are likely due to the ability of HSCT to cross the blood-brain barrier (BBB)[13]. However, HSCT treatment is considered to predict better clinical outcomes when administered at an earlier age[12]. Nevertheless, this does not necessarily result in the prevention of psychosis during adulthood, as this is more likely to develop with age, but there have been reports of altering the course of the rapid neurological disease progression in infantile Globoid Cell Leukodystrophy (GLD)[119], which may be applied to AM. Despite these reports, there is no evidence to suggest that HSCT has any significant long-term effects on the outcomes of patient neurological function and psychiatric state post-treatment and its effects on their long-term development, nor are there many studies using an adult cohort. It is, therefore, prudent that further study should be carried out into the long-term HSCT in patients with AM during adulthood, as this is when psychosis and neurological dysfunction are most likely to develop.
ERT is the most commonly used therapy in the treatment of LSDs. ERT is specifically used in the long-term treatment of adults, adolescents, and children with mild to moderate AM disease using velmanase alfa (VA), also known as Lamzede, which is known to not cross the BBB, similar to all other recombinant lysosomal enzymes used in ERT[118]. There are studies of ERT using VA in the treatment of AM, with many reports of improved clinical symptoms and quality of life post-treatment due to the reduction in mannose-rich oligosaccharide levels in bodily tissues, thereby altering disease progression and abating clinical complications. There have been reports of reduction in pain and disability in AM patients post-ERT[120], as well as patients showing improved scores in motor proficiency and increased skill acquisition in comparison to healthy peers after receiving ERT therapy[121]. This may potentially improve the psychiatry of patients by positively influencing their behavior as a consequence of their pain improvement. Treatment with VA has shown clinical benefit that was maintained for up to 4 years in patients with AM[122]. This long-term follow-up is specifically important in relation to neurocognitive decline, in which there is a need for the involvement of a neuropsychiatrist, neurologist, and neuropsychologist. However, the limitation of these studies discussed is the lack of investigation into evidence of neurocognitive improvement post-ERT, with the outcomes of these trials focusing mainly on the effects on mobility, ataxia, and respiratory dysfunction. This is because, unlike HSCT, ERT does not cross the BBB[12] and instead targets the periphery and soft tissues, therefore making it more difficult to assess whether ERT has any effect on the neurological function of patients with AM.
Potential new therapeutic developments in AM
CoQ10 analog and uncouplers
In view of the suggested involvement of mitochondrial dysfunction in the pathophysiology of AM , the use of therapeutic strategies that target the mitochondria and decrease cellular oxidative stress may be judicious. Coenzyme Q10 (CoQ10) may be an appropriate candidate therapy to consider in view of its ability to increase mitochondrial respiratory chain (MRC) activity in addition to its antioxidant capacity to decrease cellular oxidative stress[123]. Furthermore, cerebral ataxia, which is a characteristic clinical feature of AM, is commonly associated with a CoQ10 deficiency, and therefore, it may be appropriate to determine the endogenous status of this quinone prior to supplementation with exogenous CoQ10[123]. However, the ability of CoQ10 to cross the BBB is as yet uncertain, and therefore, the short-chain analog of CoQ10, idebenone, which is able to cross the BBB, may be more suitable for the treatment of cerebral mitochondrial dysfunction[123]. Another synthetic analog of CoQ10, EPI-743, has shown some therapeutic efficacy in the treatment of patients with MRC dysfunction due to its ability to cross the BBB and restore the level of the cellular antioxidant, GSH[123]. The restoration of cellular GSH levels is thought to protect the enzymes of MRC from oxidative stress-induced dysfunction, preventing further loss of function[123]. Mitochondrial uncouplers such as dinitrophenol (DNP), which can cross the BBB, may be considered to target the increased ROS associated with AM[113,124]. Uncoupling the mitochondrion decreases ROS generation through a number of mechanisms, such as by lowering the amount of oxygen in the mitochondrion and, therefore, the availability of oxygen for one-electron reduction by the MRC, which can generate the ROS, superoxide[125]. The extent to which mitochondrial dysfunction contributes to the cognitive impairment and/or psychosis reported in AM remains to be elucidated, although these conditions have been previously reported in primary mitochondrial disease patients[125]. However, it has been mentioned that secondary mitochondrial dysfunction may impact synaptic transmission and plasticity, as the mitochondria have several major functions in neurons, including the regulation of Ca2+, redox signaling, and synaptic development and plasticity[126] [Figure 3]. The inclusion of candidate therapies that target both mitochondrial dysfunction and ameliorate oxidative stress may have some potential benefits for patients if included in the treatment regime of patients from the early stages of the disease.
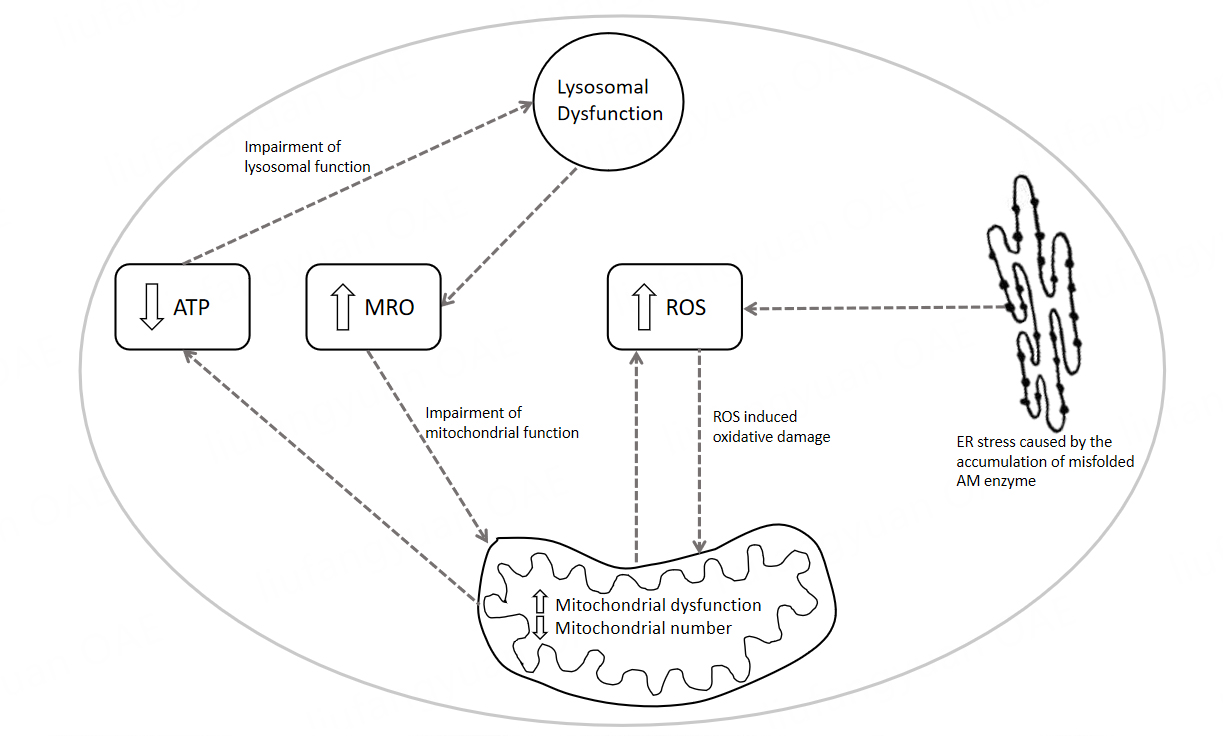
Figure 3. A summary of the potential cellular mechanisms of secondary mitochondrial dysfunction in LSDs/Alpha-Mannosidosis. The diagram depicted illustrates the putative cellular mechanism of secondary mitochondrial dysfunction in AM and other LSDs. RER: Rough endoplasmic reticulum; ROS: reactive oxygen species; AM enzyme: Alpha-Mannosidase enzyme; MRO: mannose-rich oligosaccharides; ATP: adenosine triphosphate.
CONCLUSION
Many different LSDs present with psychosis and neurological dysfunction, including patients with AM. These clinical manifestations are still poorly understood within AM disease presentation, and AM requires further elucidation into its pathophysiology, mechanisms, and long-term outcomes in relation to its neurologic and psychiatric presentation. There have been some clinical trials into the effectiveness of HSCT and ERT in AM patients, although the latter has no reported evidence of any neurologic improvement in these patients due to the inability of ERT to cross the BBB. HSCT appears to be the better option for AM treatment, particularly for the amelioration of neurocognitive symptoms and prevention of further neurodegenerative decline, despite the lack of clinical data to show improvement and/or prevention of psychosis and neurocognitive dysfunction in adult AM patients.
DECLARATIONS
Acknowledgments
The authors would like to thank all the patients with Alpha Mannosidosis.
Authors’ contributions
Study concept and design: Dewsbury MR, Hargreaves IP, Stepien KM
Critical review of the first draft: Morgan HM
Literature search and wrote the first draft of the manuscript writing: Dewsbury MR
Manuscript editing: Dewsbury MR, Hargreaves IP, Morgan HM, Stepien KM
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Stepien KM is a CI for Alpha Mannosidosis Registry. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Fuller M, Meikle PJ, Hopwood JJ. Epidemiology of lysosomal storage diseases: an overview. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 2. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11603/ [Last accessed on 14 Mar 2024].
3. Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 2012;199:723-34.
4. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281:249-54.
6. Riise Stensland HM, Frantzen G, Kuokkanen E, et al. amamutdb.no: a relational database for MAN2B1 allelic variants that compiles genotypes, clinical phenotypes, and biochemical and structural data of mutant MAN2B1 in α-mannosidosis. Hum Mutat 2015;36:581-6.
7. Stepien KM, Roncaroli F, Turton N, et al. Mechanisms of mitochondrial dysfunction in lysosomal storage disorders: a review. J Clin Med 2020;9:2596.
8. Borgwardt L, Stensland HM, Olsen KJ, et al. Alpha-mannosidosis: correlation between phenotype, genotype and mutant MAN2B1 subcellular localisation. Orphanet J Rare Dis 2015;10:70.
9. Stepien KM, Cufflin N, Donald A, Jones S, Church H, Hargreaves IP. Secondary mitochondrial dysfunction as a cause of neurodegenerative dysfunction in lysosomal storage diseases and an overview of potential therapies. Int J Mol Sci 2022;23:10573.
10. Guffon N, Tylki-Szymanska A, Borgwardt L, et al. Recognition of alpha-mannosidosis in paediatric and adult patients: presentation of a diagnostic algorithm from an international working group. Mol Genet Metab 2019;126:470-4.
11. Jakubczyk K, Dec K, Kałduńska J, Kawczuga D, Kochman J, Janda K. Reactive oxygen species - sources, functions, oxidative damage. Pol Merkur Lekarski 2020;48:124-7.
12. Tan EY, Boelens JJ, Jones SA, Wynn RF. Hematopoietic stem cell transplantation in inborn errors of metabolism. Front Pediatr 2019;7:433.
13. Adam J, Malone R, Lloyd S, Lee J, Hendriksz CJ, Ramaswami U. Disease progression of alpha-mannosidosis and impact on patients and carers - a UK natural history survey. Mol Genet Metab Rep 2019;20:100480.
14. Edelmann MJ, Maegawa GHB. CNS-targeting therapies for lysosomal storage diseases: current advances and challenges. Front Mol Biosci 2020;7:559804.
15. Dietemann JL, Filippi de la Palavesa MM, Tranchant C, Kastler B. MR findings in mannosidosis. Neuroradiology 1990;32:485-7.
16. Lipiński P, Różdżyńska-Świątkowska A, Iwanicka-Pronicka K, Perkowska B, Pokora P, Tylki-Szymańska A. Long-term outcome of patients with alpha-mannosidosis - a single center study. Mol Genet Metab Rep 2022;30:100826.
17. Borgwardt L, Thuesen AM, Olsen KJ, Fogh J, Dali CI, Lund AM. Cognitive profile and activities of daily living: 35 patients with alpha-mannosidosis. J Inherit Metab Dis 2015;38:1119-27.
18. Hafiz S, De Jesus O. Ataxia. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Available from: https://www.ncbi.nlm.nih.gov/books/NBK562284/ [Last accessed on 13 Mar 2024].
19. Kawai H, Nishino H, Nishida Y, et al. Skeletal muscle pathology of mannosidosis in two siblings with spastic paraplegia. Acta Neuropathol 1985;68:201-4.
22. Yunis JJ, Lewandowski RC Jr, Sanfilippo SJ, Tsai MY, Foni I, Bruhl HH. Clinical manifestations of mannosidosis - a longitudinal study. Am J Med 1976;61:841-8.
23. Bach G, Kohn G, Lasch EE, et al. A new variant of mannosidosis with increased residual enzymatic activity and mild clinical manifestation. Pediatr Res 1978;12:1010-5.
24. Cathey SS, Sarasua SM, Simensen R, et al. Intellectual functioning in alpha-mannosidosis. JIMD Rep 2019;50:44-9.
25. Sedel F, Baumann N, Turpin JC, Lyon-Caen O, Saudubray JM, Cohen D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J Inherit Metab Dis 2007;30:631-41.
26. Danielsen ER, Lund AM, Thomsen C. Cerebral magnetic resonance spectroscopy demonstrates long-term effect of bone marrow transplantation in α-mannosidosis. In: Zschocke J, Gibson KM, Brown G, Morava E, Peters V, editors. JIMD reports - volume 11. Berlin: Springer Berlin Heidelberg; 2013. pp. 49-52.
27. Gutschalk A, Harting I, Cantz M, Springer C, Rohrschneider K, Meinck HM. Adult alpha-mannosidosis: clinical progression in the absence of demyelination. Neurology 2004;63:1744-6.
28. Malm D, Pantel J, Linaker OM. Psychiatric symptoms in alpha-mannosidosis. J Intellect Disabil Res 2005;49:865-71.
29. Ara JR, Mayayo E, Marzo ME, et al. Neurological impairment in alpha-mannosidosis: a longitudinal clinical and MRI study of a brother and sister. Childs Nerv Syst 1999;15:369-71.
30. Majovska J, Nestrasil I, Paulson A, et al. White matter alteration and cerebellar atrophy are hallmarks of brain MRI in alpha-mannosidosis. Mol Genet Metab 2021;132:189-97.
31. Malaquias MJ, Pinto E, Oliveira J, Freixo JP, Caseiro C, Magalhães M. Alpha-mannosidosis: a novel cause of bilateral thalami and dentate nuclei hyperintensity. Can J Neurol Sci 2022;49:704-5.
32. Borgwardt L, Danielsen ER, Thomsen C, et al. Alpha-mannosidosis: characterization of CNS pathology and correlation between CNS pathology and cognitive function. Clin Genet 2016;89:489-94.
33. Zalfa C, Verpelli C, D’Avanzo F, et al. Glial degeneration with oxidative damage drives neuronal demise in MPSII disease. Cell Death Dis 2016;7:e2331.
34. Viana GM, Priestman DA, Platt FMK, Tomatsu S, Pshezhetsky AV. Brain pathology in mucopolysaccharidoses (MPS) patients with neurological forms. J Clin Med 2020;9:396.
35. Heon-Roberts R, Nguyen ALA, Pshezhetsky AV. Molecular bases of neurodegeneration and cognitive decline, the major burden of Sanfilippo disease. J Clin Med 2020;9:344.
36. Pereira VG, Martins AM, Micheletti C, D’Almeida V. Mutational and oxidative stress analysis in patients with mucopolysaccharidosis type I undergoing enzyme replacement therapy. Clin Chim Acta 2008;387:75-9.
37. Donida B, Marchetti DP, Biancini GB, et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim Biophys Acta 2015;1852:1012-9.
38. Ayodele O, Müller K, Setayeshgar S, Alexanderian D, Yee KS. Clinical characteristics and healthcare resource utilization for patients with mucopolysaccharidosis II (MPS II) in the United States: a retrospective chart review. J Health Econ Outcomes Res 2022;9:117-27.
39. Filippon L, Vanzin CS, Biancini GB, et al. Oxidative stress in patients with mucopolysaccharidosis type II before and during enzyme replacement therapy. Mol Genet Metab 2011;103:121-7.
40. Bhalla A, Ravi R, Fang M, et al. Characterization of fluid biomarkers reveals lysosome dysfunction and neurodegeneration in neuronopathic MPS II patients. Int J Mol Sci 2020;21:5188.
41. Azambuja AS, Pimentel-Vera LN, Gonzalez EA, et al. Evidence for inflammasome activation in the brain of mucopolysaccharidosis type II mice. Metab Brain Dis 2020;35:1231-6.
42. Manzoli R. Identification and characterization of signaling and axonal migration defects in the MPS II zebrafish model. Res Padua Arch 2023;8. Available from: https://hdl.handle.net/11577/3478864 [Last accessed on 14 Mar 2024].
43. Corrêa T, Poswar F, Santos-Rebouças CB. Convergent molecular mechanisms underlying cognitive impairment in mucopolysaccharidosis type II. Metab Brain Dis 2022;37:2089-102.
44. Pshezhetsky AV. Crosstalk between 2 organelles: lysosomal storage of heparin sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type c. Rare Dis 2015;3:e1049793.
45. Montero R, Yubero D, Salgado MC, et al. Plasma coenzyme Q10 status is impaired in selected genetic conditions. Sci Rep 2019;9:793.
46. Kong W, Meng Y, Zou L, Yang G, Wang J, Shi X. Mucopolysaccharidosis III in Mainland China: natural history, clinical and molecular characteristics of 34 patients. J Pediatr Endocrinol Metab 2020;33:793-802.
47. Gerken E, Ahmad S, Rattan L, Hemsley K, Barthelson K, Lardelli M. Zebrafish models of Mucopolysaccharidosis types IIIA, B, & C show hyperactivity and changes in oligodendrocyte state. bioRxiv 2023:8.
48. Villani GR, Di Domenico C, Musella A, Cecere F, Di Napoli D, Di Natale P. Mucopolysaccharidosis IIIB: oxidative damage and cytotoxic cell involvement in the neuronal pathogenesis. Brain Res 2009;1279:99-108.
49. Vitry S, Bruyère J, Hocquemiller M, et al. Storage vesicles in neurons are related to Golgi complex alterations in mucopolysaccharidosis IIIB. Am J Pathol 2010;177:2984-99.
50. Egeland MT, Tarczyluk-Wells MM, Asmar MM, et al. Central nervous system pathology in preclinical MPS IIIB dogs reveals progressive changes in clinically relevant brain regions. Sci Rep 2020;10:20365.
51. Harm TA, Hostetter SJ, Nenninger AS, Valentine BN, Ellinwood NM, Smith JD. Temporospatial development of neuropathologic findings in a canine model of mucopolysaccharidosis IIIB. Vet Pathol 2021;58:205-22.
52. Pará C, Bose P, Bruno L, et al. Early defects in mucopolysaccharidosis type IIIC disrupt excitatory synaptic transmission. JCI Insight 2021;6:e142073.
53. Martins C, Hůlková H, Dridi L, et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015;138:336-55.
54. Borges MS, Aquino MB, Vagnini L, Carneiro ZA, Fonseca JH, Lourenco CM. Lysosomal acid lipase deficiency across ages: unraveling the clinical spectrum of an under-recognized genetic disorder. Mol Genet Metab 2020;129:S32-3.
55. Pablo-Latorre R, Saide A, Polishhuck EV, Nusco E, Fraldi A, Ballabio A. Impaired parkin-mediated mitochondrial targeting to autophagosomes differentially contributes to tissue pathology in lysosomal storage diseases. Hum Mol Genet 2012;21:1770-81.
56. Settembre C, Fraldi A, Jahreiss L, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 2008;17:119-29.
57. Zhong XZ, Sun X, Cao Q, Dong G, Schiffmann R, Dong XP. BK channel agonist represents a potential therapeutic approach for lysosomal storage diseases. Sci Rep 2016;6:33684.
58. Ginzburg L, Futerman AH. Defective calcium homeostasis in the cerebellum in a mouse model of Niemann-Pick A disease. J Neurochem 2005;95:1619-28.
59. Pressey SN, Smith DA, Wong AM, Platt FM, Cooper JD. Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol Dis 2012;45:1086-100.
60. Sarkar S, Carroll B, Buganim Y, et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep 2013;5:1302-15.
61. Shen D, Wang X, Li X, et al. Lipid storage disorders block lysosomal trafficking by inhibiting TRP channel and calcium release. Nat Commun 2012;3:731-51.
62. Sun X, Marks DL, Park WD, et al. Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am J Hum Genet 2001;68:1361-72.
63. Lloyd-Evans E, Platt FM. Lysosomal Ca2+ homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011;50:200-5.
64. Potter GB, Petryniak MA. Neuroimmune mechanisms in Krabbe’s disease. J Neurosci Res 2016;94:1341-8.
65. Fiorenza MT, Moro E, Erickson RP. The pathogenesis of lysosomal storage disorders: beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum Mol Genet 2018;27:R119-29.
66. Cougnoux A, Drummond RA, Fellmeth M, et al. Unique molecular signature in mucolipidosis type IV microglia. J Neuroinflamm 2019;16:276.
67. Schedin S, Sindelar PJ, Pentchev P, Brunk U, Dallner G. Peroxisomal impairment in Niemann-Pick type C disease. J Biol Chem 1997;272:6245-51.
68. Kennedy BE, LeBlanc VG, Mailman TM, et al. Pre-symptomatic activation of antioxidant responses and alterations in glucose and pyruvate metabolism in Niemann-Pick type C1-deficient murine brain. PLoS ONE 2013;8:e82685.
69. Osellame LD, Rahim AA, Hargreaves IP, et al. Mitochondria and quality control defects in amouse model of Gaucher disease-links to Parkinson’s disease. Cell Metab 2013;17:941-53.
70. Dasgupta N, Xu YH, Li R, et al. Neuronopathic Gaucher disease: dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum Mol Genet 2015;24:7031-48.
71. Xu YH, Xu K, Sun Y, et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum Mol Genet 2014;23:3943-57.
72. Tullo MG, Cerulli Irelli E, Caramia F, et al. The spectrum of neurological and sensory abnormalities in Gaucher disease patients: a multidisciplinary study (SENOPRO). Int J Mol Sci 2023;24:8844.
73. Kartha RV, Terluk MR, Brown R, et al. Patients with Gaucher disease display systemic oxidative stress dependent on therapy status. Mol Genet Metab Rep 2020;25:100667.
74. Zhang Z, Wang X, Lin Y, Pan D. A multifaceted evaluation of microgliosis and differential cellular dysregulation of mammalian target of rapamycin signaling in neuronopathic Gaucher disease. Front Mol Neurosci 2022;15:944883.
75. Yañez MJ, Campos F, Marín T, et al. c-Abl activates RIPK3 signaling in Gaucher disease. Biochim Biophys Acta Mol Basis Dis 2021;1867:166089.
76. Shimizu T, Schutt CR, Izumi Y, et al. Direct activation of microglia by β-glucosylceramide causes phagocytosis of neurons that exacerbates Gaucher disease. Immunity 2023;56:307-19.e8.
77. Srikanth MP, Feldman RA. Elevated Dkk1 mediates downregulation of the canonical Wnt pathway and lysosomal loss in an iPSC model of neuronopathic Gaucher disease. Biomolecules 2020;10:1630.
78. Baden P, Perez MJ, Raji H, et al. Glucocerebrosidase is imported into mitochondria and preserves complex I integrity and energy metabolism. Nat Commun 2023;14:1930.
79. Teixeira CA, Miranda CO, Sousa VF, et al. Early axonal loss accompanied by impaired endocytosis, abnormal axonal transport, and decreased microtubule stability occur in the model of Krabbe’s disease. Neurobiol Dis 2014;66:92-103.
80. Singh I, Singh AK, Contreras MA. Peroxisomal dysfunction in inflammatory childhood white matter disorders: an unexpected contributor to neuropathology. J Child Neurol 2009;24:1147-57.
81. Haq E, Contreras MA, Giri S, Singh I, Singh AK. Dysfunction of peroxisomes in twitcher mice brain: a possible mechanism of psychosine-induced disease. Biochem Biophys Res Commun 2006;343:229-38.
82. Voccoli V, Tonazzini I, Signore G, Caleo M, Cecchini M. Role of extracellular calcium and mitochondrial oxygen species in psychosine-induced oligodendrocyte cell death. Cell Death Dis 2014;5:e1529.
83. Wu L, Liao X, Yang S, Gan S. Krabbe disease associated with mitochondrial dysfunction in a chinese family. Front Neurol 2021;12:750095.
84. Kreher C, Favret J, Weinstock NI, et al. Neuron-specific ablation of the Krabbe disease gene galactosylceramidase in mice results in neurodegeneration. PLoS Biol 2022;20:e3001661.
85. Hatton C, Ghanem SS, Koss DJ, et al. International DLB Genetics Consortium. Prion-like α-synuclein pathology in the brain of infants with Krabbe disease. Brain 2022;145:1257-63.
86. Karabelas AB, Walkley SU. Altered patterns of evoked synaptic activity in cortical pyramidal neurons in feline ganglioside storage disease. Brain Res 1985;339:329-36.
87. Purpura DP, Highstein SM, Karabelas AB, Walkley SU. Intracellular recording and HRP-staining of cortical neurons in feline ganglioside storage disease. Brain Res 1980;181:446-9.
88. Utz JR, Crutcher T, Schneider J, Sorgen P, Whitley CB. Biomarkers of central nervous system inflammation in infantile and juvenile gangliosidoses. Mol Genet Metab 2015;114:274-80.
89. Sano R, Annunziata I, Patterson A, et al. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca2+-dependent mitochondrial apoptosis. Mol Cell 2009;36:500-11.
90. Takamura A, Higaki K, Kajimaki K, et al. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem Biophys Res Commun 2008;367:616-22.
91. Liu S, Feng Y, Huang Y, et al. A GM1 gangliosidosis mutant mouse model exhibits activated microglia and disturbed autophagy. Exp Biol Med 2021;246:1330-41.
92. Demir SA, Timur ZK, Ateş N, Martínez LA, Seyrantepe V. GM2 ganglioside accumulation causes neuroinflammation and behavioral alterations in a mouse model of early onset Tay-Sachs disease. J Neuroinflamm 2020;17:277.
93. Gadoth N. Neuropsychiatry in late onset Tay-Sachs disease. In: Costa LV, Oliveira S, editors. Communicating rare diseases and disorders in the digital age. Hershey: IGI Global; 2020. pp. 274-91.
94. Micsenyi MC, Dobrenis K, Stephney G, et al. Neuropathology of the Mcoln1-/- knockout mouse model of mucolipidosis type IV. J Neuropathol Exp Neurol 2009;68:125-35.
95. Jezela-Stanek A, Ciara E, Stepien KM. Neuropathophysiology, genetic profile, and clinical manifestation of mucolipidosis IV-a review and case series. Int J Mol Sci 2020;21:4564.
96. Misko A, Wood L, Kiselyov K, Slaugenhaupt S, Grishchuk Y. Progress in elucidating pathophysiology of mucolipidosis IV. Neurosci Lett 2021;755:135944.
97. Mepyans M, Andrzejczuk L, Sosa J, et al. Early evidence of delayed oligodendrocyte maturation in the mouse model of mucolipidosis type IV. Dis Model Mech 2020;13:dmm044230.
98. Peng W, Wong YC, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial Ca2+ dynamics via lysosomal TRPML1. Proc Natl Acad Sci USA 2020;117:19266-75.
99. Nelson MP, Tse TE, O’Quinn DB, et al. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol Commun 2014;2:1-15.
100. Groh J, Ribechini E, Stadler D, Schilling T, Lutz MB, Martini R. Sialoadhesin promotes neuroinflammation-related disease progression in two mouse models of CLN disease. Glia 2016;64:792-809.
101. Uusi-Rauva K, Blom T, von Schantz-Fant C, Blom T, Jalanko A, Kyttälä A. Induced pluripotent stem cells derived from a CLN5 patient manifest phenotypic characteristics of neuronal ceroid lipofuscinoses. Int J Mol Sci 2017;18:955.
102. Kolikova J, Afzalov R, Surin A, Lehesjoki AE, Khiroug L. Deficient mitochondrial Ca2+ buffering in the Cln8mnd mouse model of neuronal ceroid lipofuscinosis. Cell Calcium 2011;50:491-501.
103. Dozières-Puyravel B, Nasser H, Elmaleh-Bergès M, et al. Paediatric-onset neuronal ceroid lipofuscinosis: first symptoms and presentation at diagnosis. Dev Med Child Neurol 2020;62:528-30.
104. Naseri N, Sharma M, Velinov M. Autosomal dominant neuronal ceroid lipofuscinosis: Clinical features and molecular basis. Clin Genet 2021;99:111-8.
105. Lopez-Fabuel I, Garcia-Macia M, Buondelmonte C, et al. Aberrant upregulation of the glycolytic enzyme PFKFB3 in CLN7 neuronal ceroid lipofuscinosis. Nat Commun 2022;13:536.
106. Pesaola F, Quassollo G, Venier AC, De Paul AL, Noher I, Bisbal M. The neuronal ceroid lipofuscinosis-related protein CLN8 regulates endo-lysosomal dynamics and dendritic morphology. Biol Cell 2021;113:419-37.
107. Wang Y, Wang H, Wang C. Lysosomal dysfunction, autophagic defects, and CLN5 accumulation underlie the pathogenesis of KCTD7-mutated neuronal ceroid lipofuscinoses. Autophagy 2023;19:1876-8.
110. Huang Q, Zhang YF, Li LJ, et al. Adult-onset neuronal ceroid lipofuscinosis with a novel DNAJC5 mutation exhibits aberrant protein palmitoylation. Front Aging Neurosci 2022;14:829573.
111. Lim JA, Li L, Kakhlon O, Myerowitz R, Raben N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015;11:385-402.
110. van den Dorpel JJA, van der Vlugt WMC, Dremmen MHG, et al. Is the brain involved in patients with late-onset Pompe disease? J Inherit Metab Dis 2022;45:493-501.
111. Niemann S, Beck M, Seidel G, Spranger J, Vieregge P. Neurology of adult alpha-mannosidosis. J Neurol Neurosurg Psychiatry 1996;61:116-7.
112. Zoons E, de Koning TJ, Abeling NG, Tijssen MA. Neurodegeneration with brain iron accumulation on MRI: an adult case of α-mannosidosis. JIMD Rep 2012;4:99-102.
113. Brantova O, Asfaw B, Sladkova J, et al. Ultrastructural and functional abnormalities of mitochondria in cultivated fibroblasts from α-mannosidosis patients. Biologia 2009;64:394-401.
114. Ghani S, Burney S, Ul Hussain H, Abdul Wahid M, Mumtaz H. Can velmanase alfa be the next widespread potential therapy for alpha-mannosidosis? Int J Surg 2023;109:2882-5.
115. Ceccarini MR, Codini M, Conte C, et al. Alpha-mannosidosis: therapeutic strategies. Int J Mol Sci 2018;19:1500.
116. Walkley SU, Thrall MA, Dobrenis K, et al. Bone marrow transplantation corrects the enzyme defect in neurons of the central nervous system in a lysosomal storage disease. Proc Natl Acad Sci USA 1994;91:2970-4.
117. Grewal SS, Shapiro EG, Krivit W, et al. Effective treatment of alpha-mannosidosis by allogeneic hematopoietic stem cell transplantation. J Pediatr 2004;144:569-73.
118. Mynarek M, Tolar J, Albert MH, et al. Allogeneic hematopoietic SCT for alpha-mannosidosis: an analysis of 17 patients. Bone Marrow Transplant 2012;47:352-9.
119. Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med 2005;352:2069-81.
120. Borgwardt L, Guffon N, Amraoui Y, et al. Health related quality of life, disability, and pain in alpha mannosidosis: long-term data of enzyme replacement therapy with velmanase alfa (human recombinant alpha mannosidase). J Inborn Errors Metab Screen 2018;6:232640981879685.
121. Phillips D, Hennermann JB, Tylki-Szymanska A, et al. Use of the Bruininks-Oseretsky test of motor proficiency (BOT-2) to assess efficacy of velmanase alfa as enzyme therapy for alpha-mannosidosis. Mol Genet Metab Rep 2020;23:100586.
122. Lund AM, Borgwardt L, Cattaneo F, et al. Comprehensive long-term efficacy and safety of recombinant human alpha-mannosidase (velmanase alfa) treatment in patients with alpha-mannosidosis. J Inherit Metab Dis 2018;41:1225-33.
123. Hargreaves IP. Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol 2014;49:105-11.
124. da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell 2008;7:552-60.
125. Anglin RE, Garside SL, Tarnopolsky MA, Mazurek MF, Rosebush PI. The psychiatric manifestations of mitochondrial disorders: a case and review of the literature. J Clin Psychiatry 2012;73:506-12.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Dewsbury MR, Hargreaves IP, Morgan HM, Stepien KM. Molecular basis of neurocognitive dysfunction and psychosis in Alpha-Mannosidosis. J Transl Genet Genom 2024;8:85-101. http://dx.doi.org/10.20517/jtgg.2023.58
AMA Style
Dewsbury MR, Hargreaves IP, Morgan HM, Stepien KM. Molecular basis of neurocognitive dysfunction and psychosis in Alpha-Mannosidosis. Journal of Translational Genetics and Genomics. 2024; 8(2): 85-101. http://dx.doi.org/10.20517/jtgg.2023.58
Chicago/Turabian Style
Dewsbury, Mollie R., Iain P. Hargreaves, Heather M. Morgan, Karolina M. Stepien. 2024. "Molecular basis of neurocognitive dysfunction and psychosis in Alpha-Mannosidosis" Journal of Translational Genetics and Genomics. 8, no.2: 85-101. http://dx.doi.org/10.20517/jtgg.2023.58
ACS Style
Dewsbury, MR.; Hargreaves IP.; Morgan HM.; Stepien KM. Molecular basis of neurocognitive dysfunction and psychosis in Alpha-Mannosidosis. J. Transl. Genet. Genom. 2024, 8, 85-101. http://dx.doi.org/10.20517/jtgg.2023.58
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.